De Amerikaanse medicijnautoriteit FDA heeft goedkeuring verleend aan het geneesmiddel roflumilast voor de behandeling van seborroïsch eczeem bij personen vanaf 9 jaar. Het middel, dat als een topisch schuim wordt toegediend en dat wordt geproduceerd door Arcutis Biotherapeutics onder de merknaam Zoryve, komt uit de STRATUM klinische studie naar voren als een effectief, veilig en goed verdragen steroïdevrij product voor dagelijks gebruik op alle aangetaste delen van het lichaam, inclusief behaarde gebieden. Het vertoont snelle ziekteklaring en aanzienlijke jeukvermindering, met opmerkelijke resultaten in klinische onderzoeken. Roflumilast is het eerste geneesmiddel in meer dan twee decennia dat is goedgekeurd voor seborroïsch eczeem met een nieuw werkingsmechanisme.
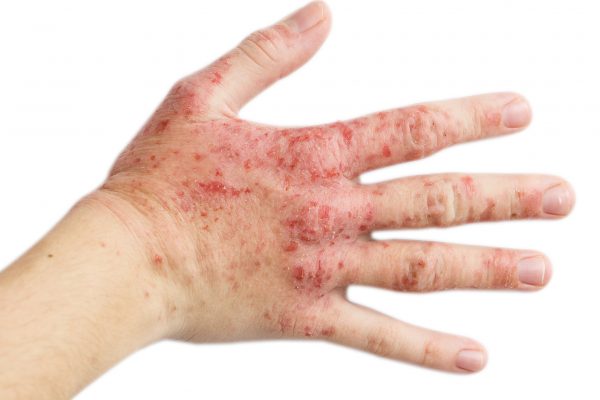
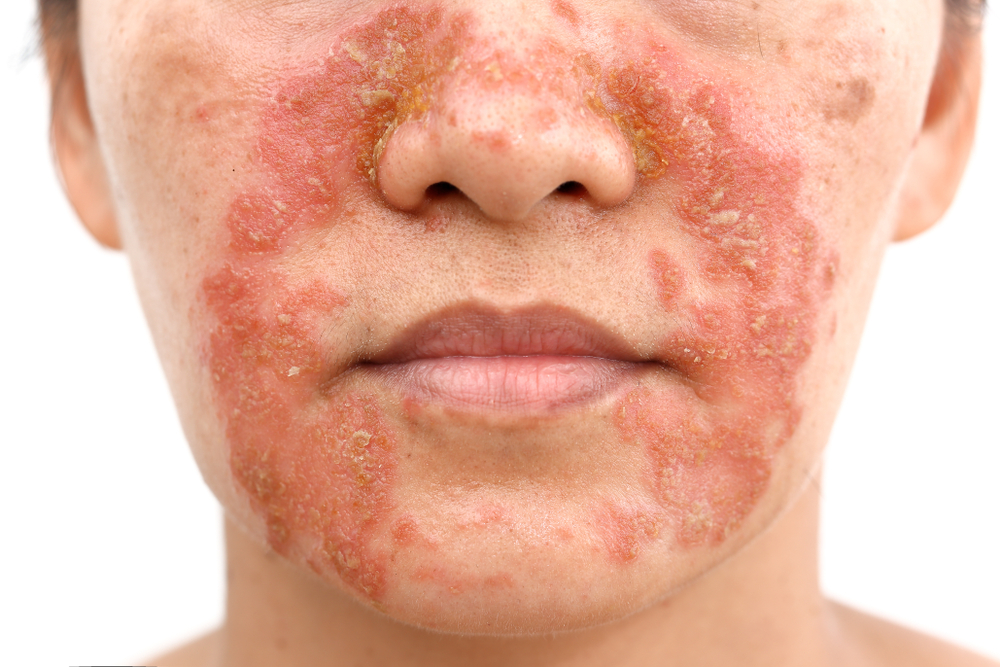
Seborroïsch eczeem, een chronische huidziekte, treft ongeveer 3% van alle mensen in Nederland, en wordt vaak geassocieerd met verminderde kwaliteit van leven en psychosociale last. Opmerkelijk is daarnaast dat ongeveer 85% van de personen met AIDS aan deze huidaandoening lijdt. Seborroïsch eczeem manifesteert zich als rode plekken bedekt met vettige schubben en intense jeuk, en met een negatieve impact op het dagelijks leven.
Positieve resultaten van fase 2 en fase 3 studies liggen aan de basis voor de goedkeuring van het roflumilast-schuim. In de STRATUM-studie behaalde 79,5% van de behandelde proefpersonen IGA-succes na 8 weken, vergeleken met 58,0% in de placebogroep (P<0.0001). In Trial 203 bereikte 73,1% van degenen behandeld met het schuim IGA-succes, vergeleken met 40,8% in de placebogroep (P<0.0001). IGA-succes werd gedefinieerd als een IGA-score van “clear” (0) of “almost clear” (1), plus een 2-graden IGA-scoreverbetering vanaf de basislijn op week 8.
Verbetering met het schuim werd al vroeg gezien. Roflumilast liet een statistisch significante verbetering zien ten opzichte van placebo op IGA-succes op week 2, het eerste beoordelingspunt in STRATUM. Bovendien bereikte 50,6% van degenen in de schuimgroep complete klaring (IGA=0) op week 8.
De STRATUM-studie toonde ook statistisch significante verbetering ten opzichte van placebo op alle secundaire eindpunten, inclusief jeuk, schilfering en erytheem (roodheid). Meer dan 60% van de proefpersonen bereikte een ≥4-punt vermindering in jeuk op week 8, gemeten met de Worst Itch-Numerical Rating Score (62,8% roflumilast vs. 40,6% placebo; P=0,0001). Er werden ook significante verbeteringen in jeuk gerapporteerd op week 2 en week 4. Proefpersonen behandeld met het schuim meldden een 28% verbetering in jeuk vanaf aanvang studie in 48 uur (vergeleken met 13% in de placebo-groep, P=0,0024).
Bovendien bereikte meer dan 50% van degenen behandeld met het schuim een erytheem (roodheid) score van 0, en meer dan 50% bereikte een schilfering score van 0 op week 8. Behandeling met het schuim toonde een aanzienlijk grotere verbetering in door patiënten gerapporteerde resultaten al op week 2, gemeten via de Dermatology Life Quality Index (DLQI), met verbeteringen die behouden bleven tot week 8.
Het roflumilast-schuim werd goed verdragen met een gunstig veiligheids- en tolerantieprofiel gedurende maximaal 52 weken behandeling. Het incidentiecijfer van behandelingsgerelateerde bijwerkingen (‘treatment emergent adverse effects’, TEAEs) was laag en vergelijkbaar tussen actieve behandeling en placebo, waarbij de meeste TEAEs werden beoordeeld als lichte tot matig in ernst. Er waren geen behandelingsgerelateerde ernstige bijwerkingen (‘serious adverse effects’, SAEs). Over het algemeen waren de meest voorkomende bijwerkingen die optreden bij ≥1% van de proefpersonen in de gecombineerde fase 2- en fase 3-studiepopulaties nasofaryngitis (1,5%), misselijkheid (1,3%) en hoofdpijn (1,1%).
Verwacht wordt dat het geneesmiddel Zoryve al zeer snel op de markt komt in de Verenigde Staten. In Europa wordt medio 2024 een besluit tot eventuele toelating verwacht door de Europese geneesmiddelenautoriteit EMA.
Referentie
Persbericht van Arcutis aangaande de goedkeuring door de FDA